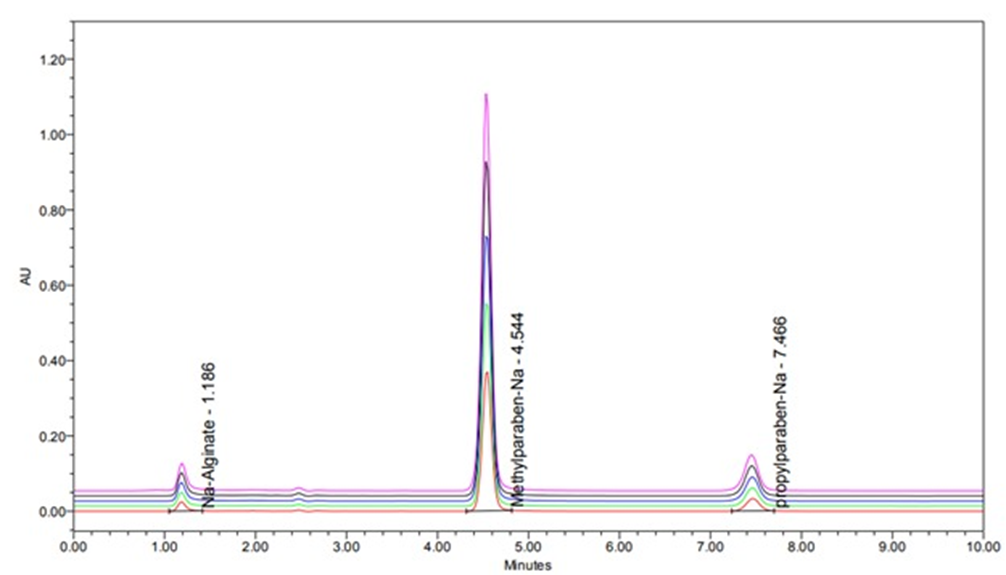
A stability-indicating high-performance liquid chromatographic (HPLC) method was developed and validated for simultaneous quantification of sodium alginate (Na Alg), sodium methylparaben (Na-MP), and sodium propylparaben (Na-PP) in oral suspension formulations. This method addresses the need for reliable quality control of complex pharmaceutical matrices. Separation was achieved on an Ultisil XB-CN(Cyanopropyl) column at 40 °C, using an isocratic mobile phase composed of 0.05 M potassium dihydrogen phosphate buffer (pH 5.2) and acetonitrile (80:20, v/v) at a flow rate of 1.5 mL/min, with detection at 210 nm. The method was validated in accordance with the ICH Q2(R1) and USP guidelines to assess linearity, accuracy, precision, specificity, and robustness. The technique demonstrated excellent linearity (R2 > 0.9998) over the concentration ranges of 10–1500, 1–150, and 0.16–24 μg/mL for Na-Alg, Na-MP, and Na-PP, respectively. The validation results showed high accuracy expressed as recovery (98.65–101.88%) across the tested concentration range of 50% to 150%, precision (%RSD < 1.9%), and sensitivity (LODs: 5.30, 0.038, and 0.062 μg/mL; LOQs: 17.65, 0.127, and 0.207 μg/mL) for Na-Alg, Na MP, and Na-PP, respectively. The degradation products were successfully resolved from the analytes, thereby confirming the specificity of the method. The developed HPLC method used to analyze 34 commercial alginate suspension samples was highly precise (%RSD < 1.5%) and accurate (94.66%–105.20% recovery). It reliably quantified Na-Alg and preservatives (Na-MP and Na-PP), with results consistently matching the label claims. This robust method is suitable for routine quality control and stability testing, and effectively combines regulatory compliance with practical pharmaceutical analysis.


![Schematic diagram of chemometrics- spectrophotometric overview [30]](https://journals.su.edu.ye/public/journals/5/submission_2481_2481_coverImage_en_US.png)